
Translate this page into:
Cutaneous vasculitis as the sole clinical feature of primary Sjögren’s syndrome
*Corresponding author: Sumit Sehgal, Department of Dermatology, Ananta Institute of Medical Sciences and Research Centre, Rajsamand, Rajasthan, India. summitsehgal934@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Agrawal S, Sehgal S, Kamboj R. Cutaneous vasculitis as the earliest and sole clinical feature in primary Sjögren’s syndrome. Indian J Skin Allergy. 2024;3:77-9. doi: 10.25259/IJSA_48_2023
Dear Editor,
Sjögren’s syndrome (SS) is an autoimmune condition commonly characterized by keratoconjunctivitis sicca, xerostomia, and various other extraglandular features such as cutaneous vasculitis (CV).[1]
CV in SS is relatively common,representing one of the most important extraglandular manifestations. CV in SS can be associated with more severe disease activity and comorbid systemic manifestations. The prevalence of CV in SS has been reported between 5% and 10%.[2] Primary SS presents alone while secondary SS occurs in association with other autoimmune diseases. The diversity of symptomatic expression in SS can sometimes cause a delay in reaching the correct diagnosis. Early diagnosis of SS is often missed, especially when extraglandular symptoms such as vasculitis lesions are the sole clinical presentation.[3]
Herein, we are discussing a case of primary SS in a middle-aged female who was being treated for repeated episodes of CV (palpable purpura) suspected secondary to HenochSchonlein purpura/urticarial vasculitis/drug-induced rash for one year.
A 43-year-old female presented to our dermatology outpatient services with numerous reddish-colored, raised lesions arranged in a net-like pattern associated with low-intensity itch for one week. The onset of the presenting lesions was associated with the prodrome of fever, malaise, and generalized weakness. Dermatological examination revealed multiple erythematous, 5–15 mm sized purpuric lesions in the retiform pattern present over both extensor and flexor aspects of the bilateral lower limbs [Figure 1 and 2]. She also reported multiple episodes of similar lesions in the past one year that resolved with the medications. In addition, there was a history of the development of multiple, tender, and nodular lesions over the flexor aspect of both ankles 3–4 weeks back that ulcerated to form the well-defined round to oval 1–2 cm sized ulcers within the following 1–2 weeks. Figure 2 shows the ulcerated lesions covered with eschars when she presented to us after 3–4 weeks after the onset of the lesions. A net-like pattern of flat erythema with normal skin color was also present over the thighs and knees [livedo reticularis]; [Figure 3]. Her medical records suggested that she had been treated as Henoch-Schonlein purpura, urticarial vasculitis, and drug-induced rash during the past relapses of the symptoms and had also been, frequently, prescribed the potent topical and systemic corticosteroids to control the flare of the symptoms. There were no signs of any digital gangrene. There was no history of any abdominal pain (intestinal angina in small-to-medium vessel vasculitis, especially polyarteritis nodosa), loss of bladder/bowel control, sensation loss/paralysis in one or more areas of the body, tingling, burning, pain, or other abnormal sensations. There was also neither history nor medical records of any weight loss, photosensitivity, arthralgia, oral ulcers, proximal muscle weakness, vision abnormalities, tightening of the skin, dry cough/recurrent upper respiratory tract infections, and recurrent oral/genital ulcers. There was no history of similar symptoms in family members. The patient denied any discomfort/grittiness in the eyes. There was also no history of dental caries, foul breath, and dryness of mouth. The chronic and recurrent nature of the purpuric lesions prompted a detailed evaluation to find out the underlying disorder, if present. Routine laboratory investigations revealed mild iron deficiency anemia. Inflammatory markers (erythrocyte sedimentation rate and C-reactive protein) were within normal range. Routine urinary examination was also within normal limits. A deep thickness incisional biopsy was taken, including the purpuric lesions over the right lower leg, and histopathological examination suggested the findings of leukocytoclastic vasculitis (LCV), as shown in [Figure 4]. Rheumatoid factor, anti-nuclear antibodies, anti-double-stranded deoxyribonucleic acid antibodies, serum complements, serum protein electrophoresis, and cryoglobulins were negative. Viral markers, including for Hepatitis C, were also negative. High-resolution computerized tomography of the chest and abdomen was normal. However, the autoimmune antibody panel test indicated that she was strongly positive for anti-Ro and anti-La antibodies. Antibody profile tests prompted us to carry out the sicca syndrome-related symptoms-specific investigations and Schirmer’s test was found positive for both eyes (right eye – 2 mm and left eye – 4 mm). However, we could not get the minor salivary gland biopsy as the patient refused for the same.
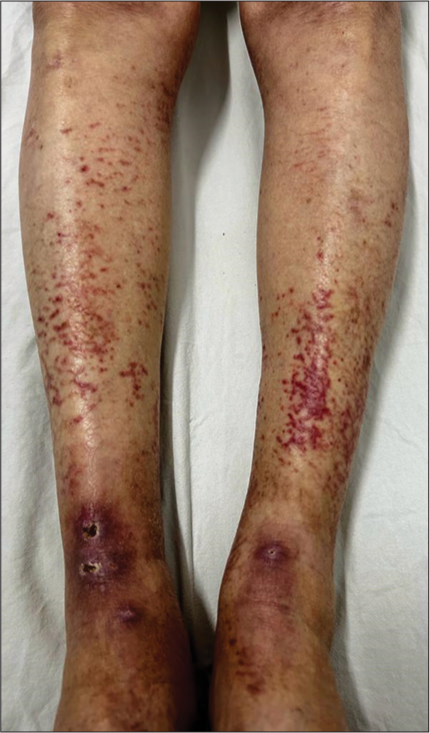
- Multiple, erythematous, and purpuric lesions in the retiform pattern present over the extensor aspect of the bilateral lower limbs.
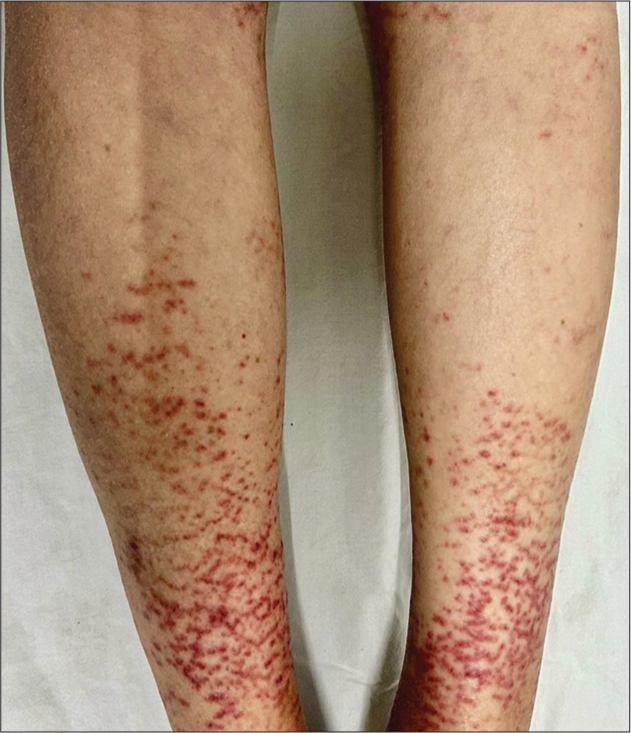
- Noduloulcerative lesions covered with eschars over dorsal aspect of the bilateral feet along with multiple, erythematous, and purpuric lesions in the retiform pattern.
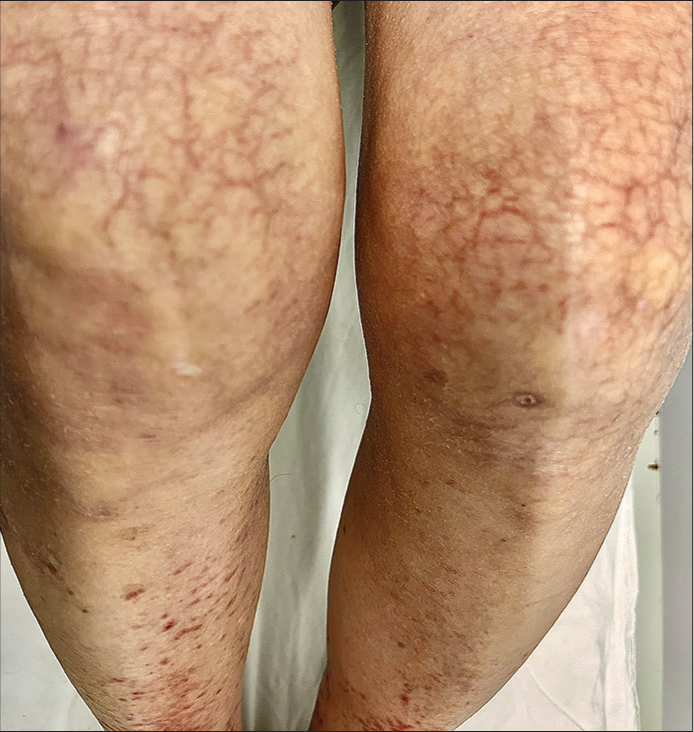
- A net-like pattern of flat erythema with normal to pale skin color in between (livedo reticularis) over bilateral thighs and knees.
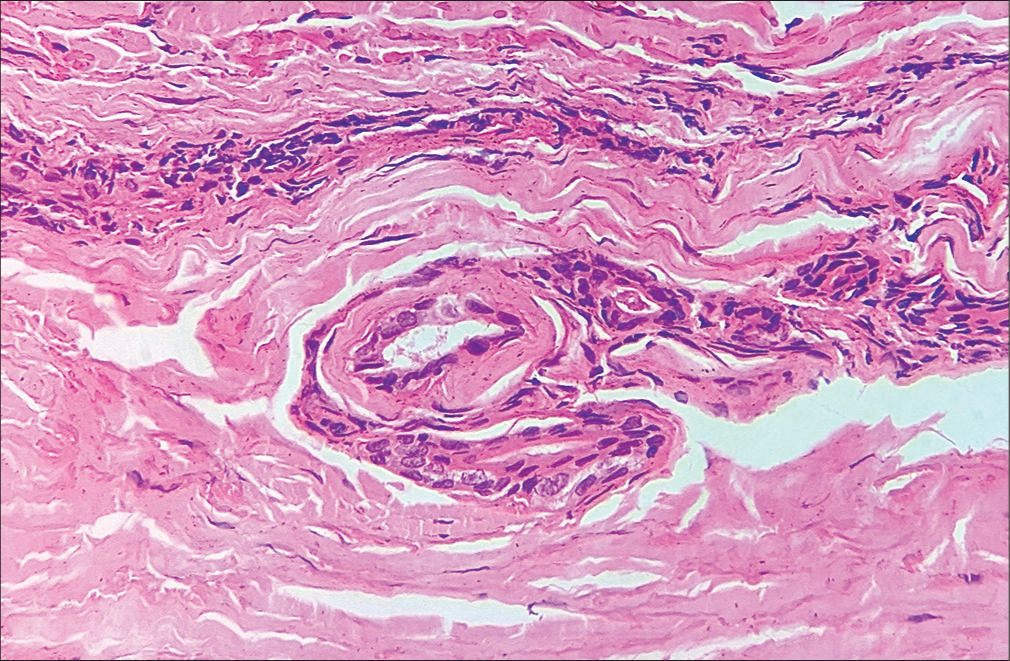
- Histopathological examination of incisional biopsy from newly developed purpuric lesions showing vessel wall destruction by the infiltration of neutrophils and fibrinoid necrosis around the vessel wall can be seen (H&E: Hematoxylin and Eosin ×40).
Based upon the positive Schirmer’s test, anti-Ro/La antibodies, purpuric-ulceronodular lesions, livedo reticularis, and absence of systemic symptoms, the final diagnosis of primary SS with small to medium vessel vasculitis (cutaneous polyarteritis nodosa) was made. Ophthalmological and rheumatology experts were consulted. Treatment was started in the form of the initial dose of oral prednisolone at 0.5 mg/kg/day with a slow tapering schedule (starting dose 30 mg/day to the stopping dose of 5 mg/day within 4 weeks). Oral hydroxychloroquine 400 mg/day in two divided doses was started alongside. Purpuric lesions resolved completely within three weeks. She was also advised for the use of artificial tears 4–5 times a day along with Omega-3 essential fatty acid supplement and moisture chamber spectacles. Chlorhexidine mouth rinse was advised at least twice a day, along with other measures to maintain good oral hygiene. At present, she is on fortnightly follow-up.
SS is a chronic autoimmune lymphoplasmacytic infiltration and progressive destruction of the salivary and lacrimal gland, leading to classical symptoms of dry mouth and dry eyes.[1,2] SS covers a broad clinical spectrum,with half of the patients presenting with variable combinations of systemic and extraglandular manifestations.[3] Although SS affects approximately 2% of the adult population,[1] it remains undiagnosed in more than half of the population.[4]
There are a great number of other symptoms which might arise before the onset of ocular and oral dryness, including skin lesions, among which vasculitis is common. CV prevalence in SS has been reported to be 5%-10%, and it can be associated with high titers of rheumatoid factor and the hallmark antibodies (anti-Ro and anti-La) in SS.[2,5] Clinically, CV manifests in a single episode or may develop a relapsing course. The most commonly seen lesions are palpable and/or non-palpable purpura in 88% of the cases, over the lower limbs. Urticarial vasculitis can develop, rarely. Ulcers can be present in up to 8% of patients.[2] Cryoglobulins are positive in 3%-4% of the patients and are associated with a higher prevalence of lymphoma.[6] The histopathologic examination of the vasculitic lesions shows LCV in 90% of cases involving the small cutaneous vessels. Direct immunofluorescence can be positive in up to 90% of cases, also. We did not carry out anti-neutrophil cytoplasmic antibody (ANCA) detection test on our patient. However, ANCA-associated vasculitis has been reported in SS, although rarely.[7]
A recent study reported that as many as 40% of SS subjects with clear objective evidence of dry eyes had no symptoms.[8] Similarly, our patient had the symptoms of recurrent LCV alone for around one year with no other symptom(s) pointing to SS. Her medical records suggested that she was not screened for the presence of underlying autoimmune disorders in the past, despite being prescribed for the high doses of systemic corticosteroids on multiple occasions. SS is usually considered an indolent systemic autoimmune disease, affecting the quality of life rather than mortality. Moreover, delayed or missed diagnosis leads to inappropriate management. More commonly, in such clinical scenarios, many physicians/dermatologists opt to only resolve the acute flare of symptoms rather than to adopt a comprehensive approach for long-term management, which eventually becomes troublesome for the patient and the doctor himself. Missed diagnosis coupled with inadequate/inappropriate treatments can also lead to unstable disease activity. Through this case, we are re-highlighting the diverse clinical spectrum of SS where extraglandular symptoms can also be the earliest and predominating to develop. It is prudent to include the SS within the rheumatologic differentials when approaching patients with new skin lesions such as recurrent purpura, nodules, and ulcers of the lower limbs, even in the absence of its classical signs and symptoms.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Sjogren's syndrome presenting with solely cutaneous features. Diagnostics (Basel). 2021;11:1260.
- [CrossRef] [PubMed] [Google Scholar]
- Cutaneous and mucosal manifestations of Sjögren's syndrome. Clin Rev Allergy Immunol. 2017;53:357-70.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical manifestations and early diagnosis of Sjögren syndrome. Arch Intern Med. 2004;164:1275-84.
- [CrossRef] [PubMed] [Google Scholar]
- Sjögren's syndrome In: Kassirer JP, Greene HL, eds. Current therapy in adult medicine (4th ed). Baltimore, MD: Mosby; 1997. p. :1291-8.
- [Google Scholar]
- Autoimmune response and target autoantigens in Sjögren's syndrome. Eur J Clin Invest. 2010;40:1026-36.
- [CrossRef] [PubMed] [Google Scholar]
- Cryoglobulinemia in Sjögren syndrome: A disease subset that links higher systemic disease activity, autoimmunity, and local B cell proliferation in mucosa-associated lymphoid tissue. J Rheumatol. 2017;44:1179-83.
- [CrossRef] [PubMed] [Google Scholar]
- Characterization of systemic disease in primary Sjögren's syndrome: EULAR-SS Task Force recommendations for articular, cutaneous, pulmonary and renal involvements. Rheumatology (Oxford). 2015;54:2230-8.
- [CrossRef] [PubMed] [Google Scholar]
- Correlations between commonly used objective signs and symptoms for the diagnosis of dry eye disease: Clinical implications. Acta Ophthalmol. 2014;92:161-6.
- [CrossRef] [PubMed] [Google Scholar]




